
芸香科柑橘属包含人们熟知的多种水果,如橘子、甜橙、柚子、柠檬、青柠等,这一成员众多且纷乱的水果家族拥有悠久的栽培历史,并且具有重要的经济价值。随着基因测序技术的不断发展,柑橘属的起源与驯化问题以及其特殊性状形成机制也开始变得明晰。
1
甜橙基因组图谱构建完成
(Nature Genetics, 2012)
华中农业大学在2012年首次完成了甜橙(Citrus sinensis)基因组构建工作,为了降低组装难度,研究人员采用双单倍体测序方法成功绘制出了甜橙的87%的基因组序列,组装基因组大小为320Mb,ContigN50为49.89Kb,ScaffoldN50为1.69Mb,编码蛋白的基因数量为29,445个。
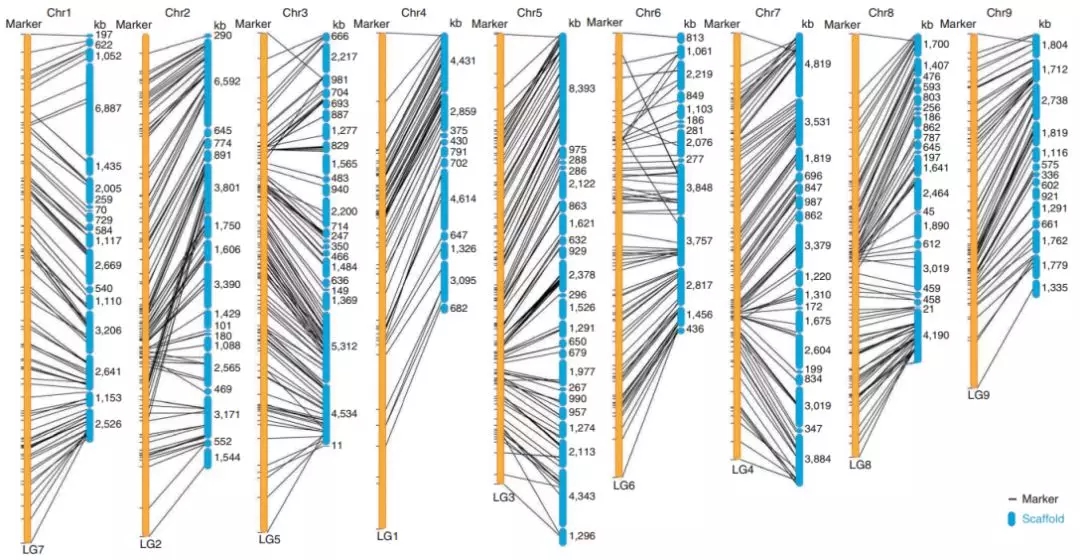
图1 组装的基因组与遗传图谱比对
重测序分析表明二倍体甜橙具有较高的杂合度(50%),同时SNPs密度偏好性发现柚和柑橘的贡献度为1:3,并有证据表明甜橙的叶绿体很可能来源于柚。因此,研究人员推断:母本柚(pummelos)与父本柑橘(mandarins)进行杂交,再与母本柚(pummelos)回交,从而形成甜橙。
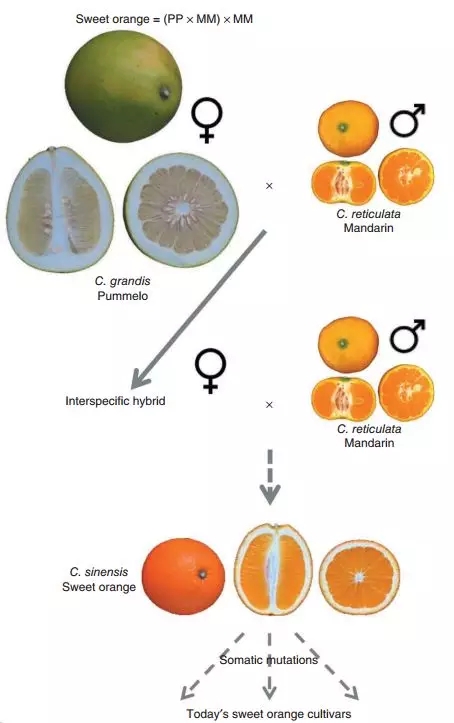
图2 甜橙起源的模型
对甜橙基因组分析,揭示甜橙基因组的进化历史,同时发现甜橙中与维生素C合成有关的基因出现基因扩增等现象,进一步结合RNA-Seq进行联合分析,发现GalUR基因表达上调很可能是甜橙富含维C的重要原因。
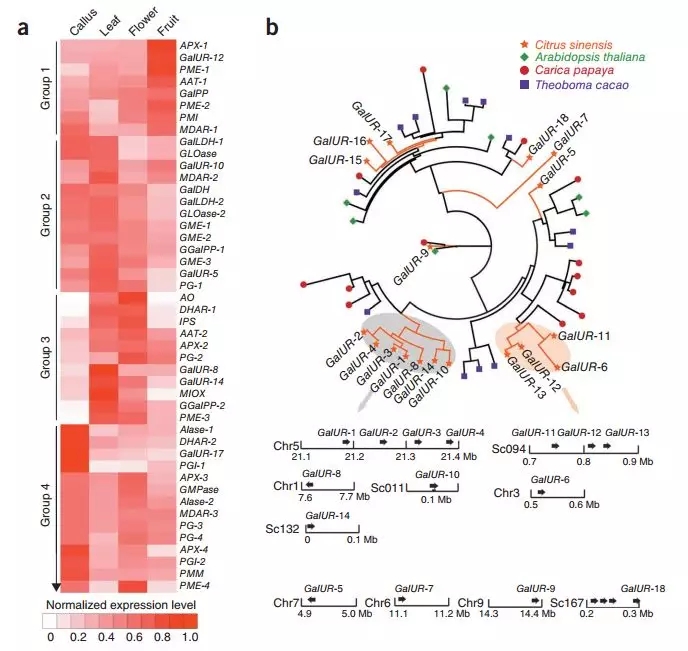
图3 与维C代谢相关的基因表达与进化
2
橘、柚、橙基因组测序初步揭示柑橘属复杂的驯化过程
(Nature Biotechnology, 2014)
2014年,美国科学家完成了几种柑橘属水果的基因组测序,其中包括四种柑橘、两种柚和酸橙,这一研究成果发表在Nature Biotechnology杂志上,该研究构建了更高质量的柑橘属物种基因组,较之前发表的甜橙基因组质量有显著提高(Contig L50=119Kb,ScaffoldL50=6.8Mb)。除此之外,该研究也对柑橘属的演化、驯化等问题进行了探讨。该研究表明,栽培柚来源祖先种C. maxima,栽培宽皮桔来源C. reticulata,并有C. maxima的渗入。
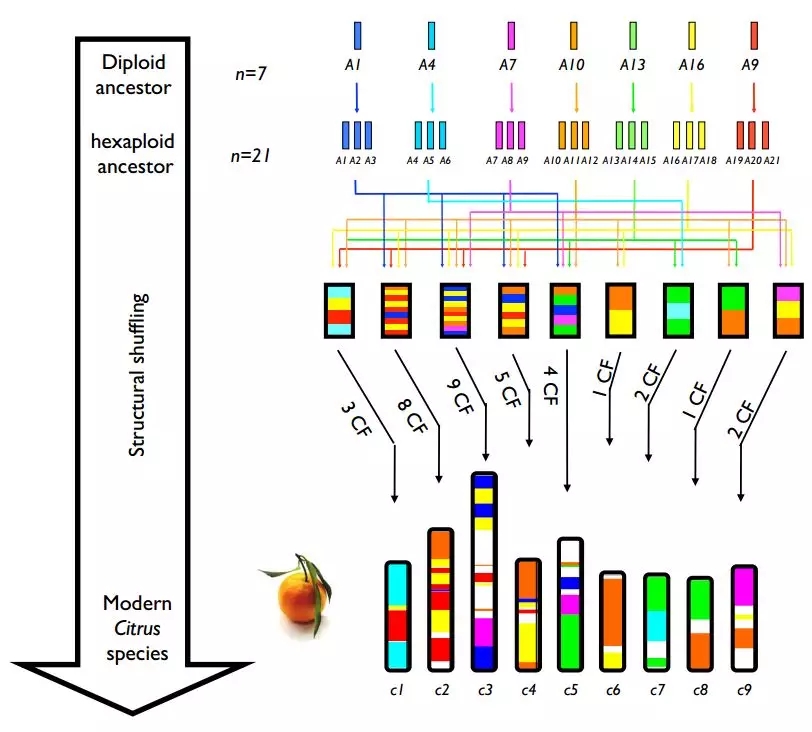
图4 柑橘属染色体进化关系
3
柑橘属基因组测序揭示其多胚性状形成机制
(Nature,2017)
继甜橙基因组公布以后,华中农业大学同一研究团队再一次完成了四个柑橘代表种的基因组构建工作,并通过联合比较基因组、重测序以及转录组等等方法解析了柑橘“多胚”形成的分子基础,锁定了关键基因CitRWP。
研究人员利用单分子测序技术(PacBio)构建了迄今为止最为完整的柑橘基因组,其中Contig N50为2.2 Mb,是已经报道的柑橘基因组的18倍以上。
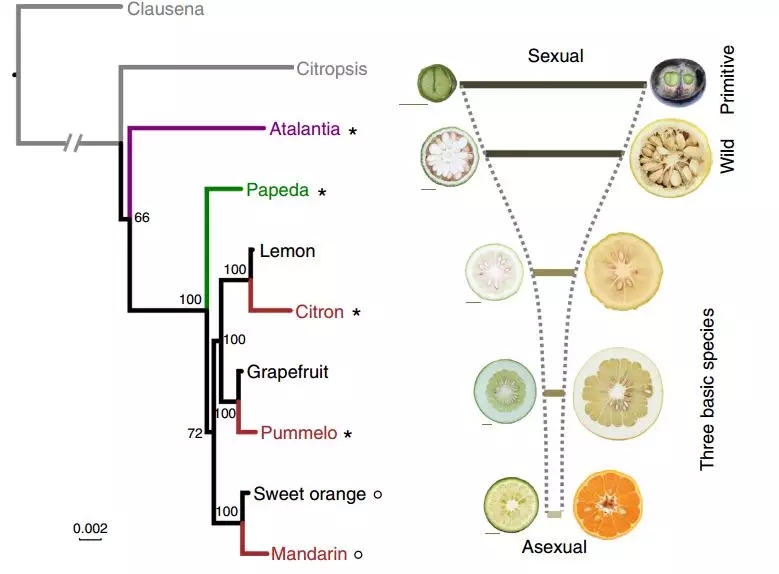
图5 柑橘属系统进化关系及性状表现
进一步对100份代表性原始、野生和栽培柑橘进行深度测序及群体比较,分析表明原始柑橘的遗传多样性最高,栽培柑橘中的生殖和能量代谢相关的基因受到了选择。
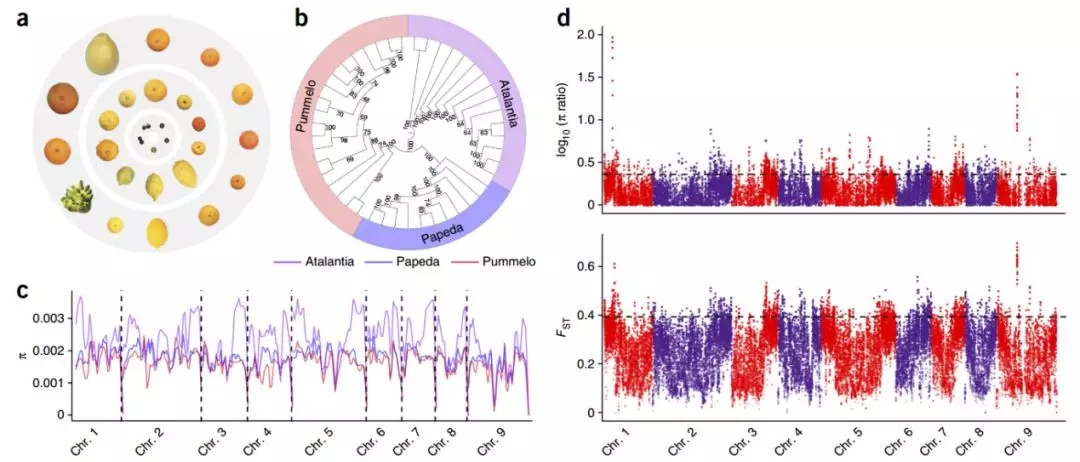
图6 柑橘遗传多样性和群体差异分析
本研究利用柑橘基因组平台优势结合前期构建的遗传群体,针对性设计了极端表型混池测序和局部基因关联分析的策略,将柑橘多胚位点定位到一段80Kb的区域,包含11个候选基因。进一步精细剖析发现候选基因CitRWP与多胚性状的关联程度最高,并表现出胚珠特异表达的特点,且在多胚的表达显著高于单胚。
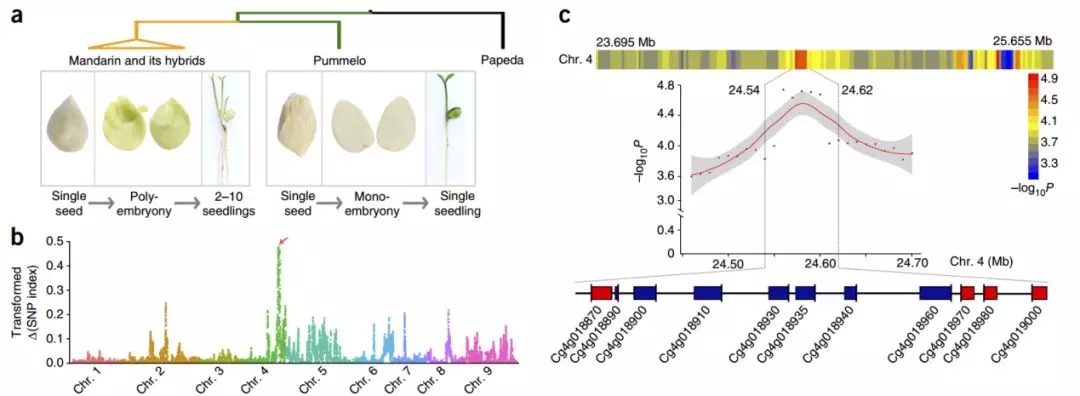
图7 柑橘多胎化基因定位
4
柑橘属泛基因组测序揭示其起源与进化机制
(Nature,2018)
该研究分析了已发表的柑橘属基因组和30种新测序的基因组(共60种材料的基因组),通过基因组学分析、系统发育分析以及生物地理学分析,揭示了柑橘属植物的起源、进化以及驯化历史。
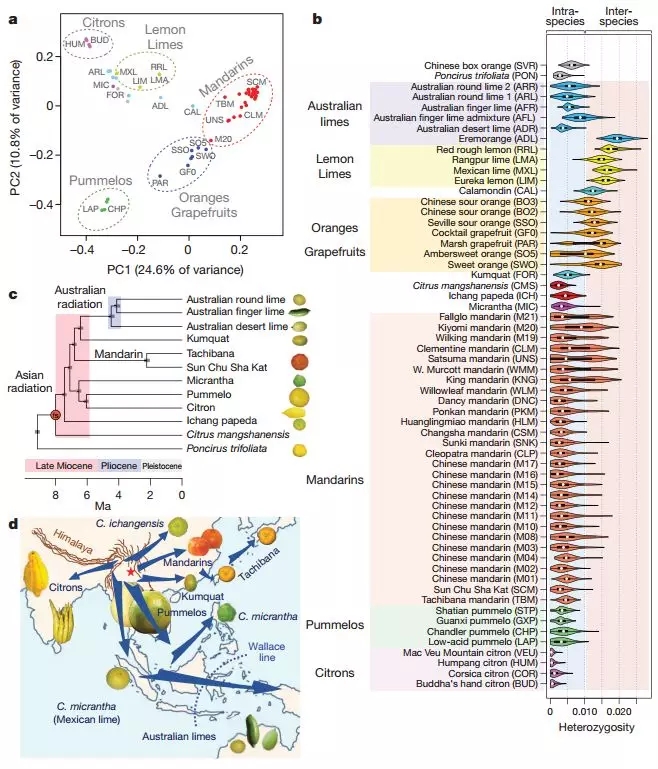
图8 柑橘属遗传结构、杂合分布及系统进化分析
基于59万个AIM(Ancestry Informative Marker or SNP),找到46份柑橘材料的片段祖先来源。在28个柑橘基因组中,23个发现有柚子基因组的贡献,进一步分析后,研究人员绘制了主要柑橘属成员的的遗传家谱。
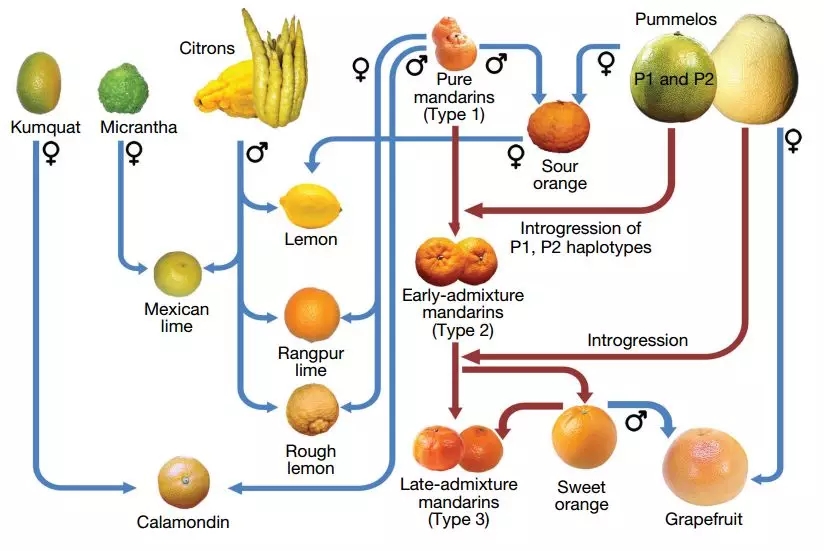
图9 主要柑橘属成员家谱图
该研究表明柚果实大小与柚基因混入比例之间呈强正相关(r=0.88)。果实风味(酸度)GWAS关联到8号染色体0.3-2.2Mb区间,在这个区域,甜柑橘在该区域有柚混入,而酸柑橘没有,最后从该区域的基因中筛选到一个调节柠檬酸合成的IDH基因。
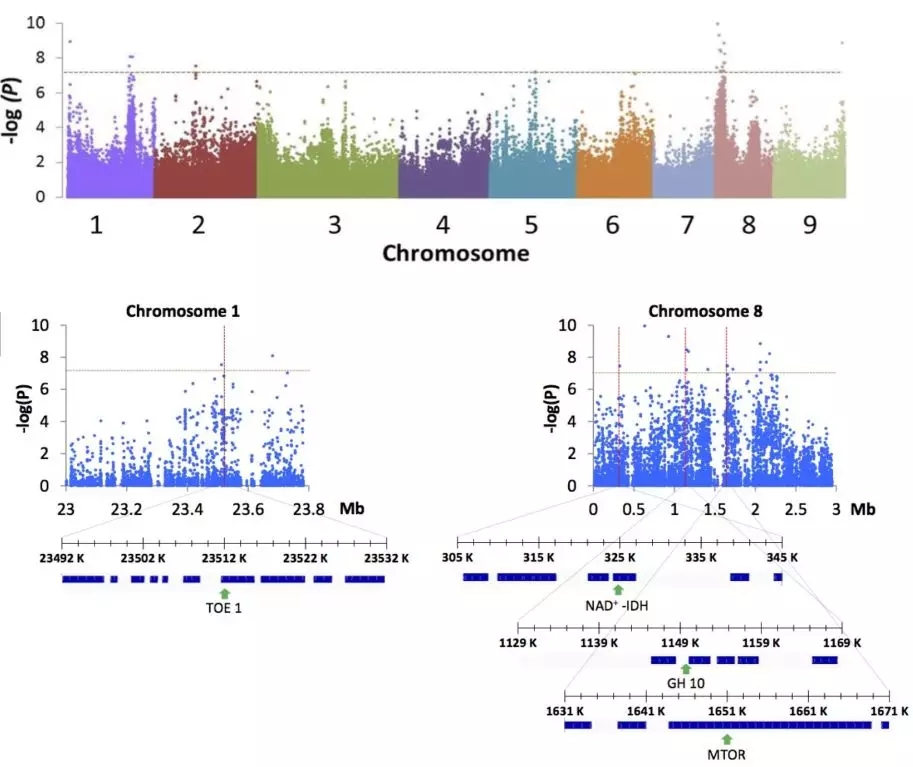
图10 GWAS分析柚果实大小及酸度相关基因定位
柑橘基因组的研究趋势与水稻和大豆基本类似,都经历了以下过程:
1.从单个基因组构建研究深入到泛基因组水平,从而能够更全面地获得该物种的所有基因资源和变异信息(小广告:如果利用10X Genomics技术,能够以超低的成本得到高质量的基因组,可谓泛基因组的构建的利器);
2.利用重测序BSA、遗传图谱、GWAS技术对所要研究的性状进行基因组定位,从而将基因与性状进行关联,如柑橘的研究中,利用BSA技术定位了柑橘多胚性状相关基因,利用GWAS定位了果实风味相关基因;
3.利用重测序群体进化分析,对物种在人工驯化或自然选择的过程中发生的性状上的定向改变和亚种形成进行过程研究,从而在基因层面解析物种的起源和进化问题;
4.利用转录组测序,从转录调控层面和表达水平来辅助生物学问题的解决。
博奥晶典作为一家致力于打造技术平台最完善的高通量技术服务机构,能够提供全面的基因组测序和分析服务。基于全新的10X Genomics平台,可以高质量地构建出动植物基因组图谱,并对所有变异位点,尤其是复杂结构变异进行全面检测;另外,依托博奥生物强大的生信分析团队和丰富的经验,能够完美完成动植物GWAS、群体遗传进化等各项大规模深度分析,为您的科研路提供强有力的支持。
动植物基因组专题 | 水稻:我们不一样,每个品种都有不同的境遇
参考文献
XuQ, Chen L L, Ruan X, et al. The draft genome of sweet orange (Citrus sinensis)[J]. Nature Genetics,2013, 45(1):59-66.
WuG A, Prochnik S, Jenkins J, et al. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication[J]. Nature Biotechnology, 2014, 32(7):656-62.
WangX, Xu Y, Zhang S, et al. Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction[J]. Nature Genetics, 2017,49(5):765-772.
WuG A, Terol J, Ibanez V, et al. Genomics of the origin and evolution of Citrus[J]. Nature, 2018.
博奥晶典科研服务事业部 李超、李媛媛 | 文案
部分配图来源于网络 侵删