PART1复制型病毒(RCR/RCL)
复制型病毒/病毒载体回复突变(Replication Competent Virus, RCV),可产生于病毒载体制造过程中的所有步骤当中。并且,RCV感染宿主细胞后能随宿主细胞在培养液中增殖、扩增对人体健康具有严重的隐患,是免疫细胞治疗类产品,如CAR-T产品的主要安全性风险之一。近年来,CAR-T细胞制备过程中,伴随载体的不断升级优化,重组产生的复制型逆转录病毒/慢病毒(Replication Competent Retrovirus / Lentivirus, RCR/RCL)逐渐降低,但产生RCR/RCL的风险隐患至今仍不能完全排除。由2022年5月发布的《体外基因修饰系统药学研究与评价技术指导原则(试行)》指出RCR/RCL检测作为安全性检测在细胞药物的研发和生产过程中至关重要,相关产品不得检出RCR/RCL,可知病毒载体相关产品中,RCR/RCL的检测至关重要。
慢病毒( lentivirus,LV) 载体是以人类免疫缺陷Ⅰ型病毒( human immunodeficiency virus,HIV-1) 为基础发展起来的基因治疗载体,它源于逆转录病毒,但又有所不同。LV与一般的逆转录病毒载体相比较,具有感染能力强、容纳更大目的片段的基因、能感染分裂细胞和非分裂细胞和很少引发免疫反应等优点,目前被选为CAR-T细胞产品的主要病毒载体。
《CAR-T细胞治疗产品质量控制检测研究及非临床研究考虑要点》一文提到:目前 RCR/RCL 的检测方法主要有指示细胞培养法、ELISA法(p24蛋白测定)、PCR/q-PCR法(通过psi-gag或VSV-G聚合酶链反应)和转录酶活性测定法(PERT)等。其中,利用指示细胞培养法对生产过程中的病毒(上清液、病毒生产终末期细胞)存在的RCL进行培养扩增,并在培养终点进行重复检测是FDA推荐的RCL检测方法。
指示细胞培养法分为2个阶段,初始扩增阶段和指示阶段:初始扩增阶段是将待测样品与C8166-45细胞(人类脐带血淋巴细胞)共培养21天,至少传代5次后收集细胞上清,分别采用ELISA法测定培养基中的P24蛋白表达量和与新的C8166-45细胞一起共培养,7 d后采用2种不同原理的方法检测RCL,如ELISA、PCR或qPCR(psi-gag/VSV-G PCR,ELISA p24,PERT)。RCL指示细胞培养法分组和检测如图1和2。
PART3RCL检测方法分析
根据现有法规和指导原则可以发现,RCL的检测方法主要包含指示细胞培养法、ELISA法、PCR/q-PCR法和PERT法。因为指示细胞培养法检测需28天,并且因为阳性对照病毒的性质,要求其需要在P3或者P2实验室环境中进行,致使该方法具有耗时长,环境要求高的特点。相比而言,基于VSV-G序列的q-PCR方法或基于psi-gag序列的PCR方法,因其具有检测时间短、环境条件简单、灵敏度高和可重复性强等优点,被许多CAR-T申报企业作为快速放行方法。
除却指示细胞培养法,其他3种快速检测RCL的检测方法优缺点如下:
01 ELISA 法(p24 蛋白测定)
ELISA 法(p24 蛋白测定):适用于由载体生产过程中病毒基因组之间的重组形成RCL的检测。但,对于VSV-G 包膜质粒和来源于其他病毒的 gal-pol 基因功能组件之间发生重组不表达p24蛋白的假型病毒不适用。其优点为适用性广(浓缩载体、终末细胞、血清血浆等多种样本)、灵敏度高和可定量等。
02 qPCR/PCR法
使用qPCR的方法测定VSV-G基因和PCR方法检测由病毒载体质粒和包装质粒重组产生的psi-gag基因序列,具有灵敏度高,可重复性和操作简单等优点。
03 PERT法
测定逆转录酶活性的方法,它可以用来检测RCL相关的不同结构的逆转病毒,缺点为在某些细胞检测中,存在背景高的现象。
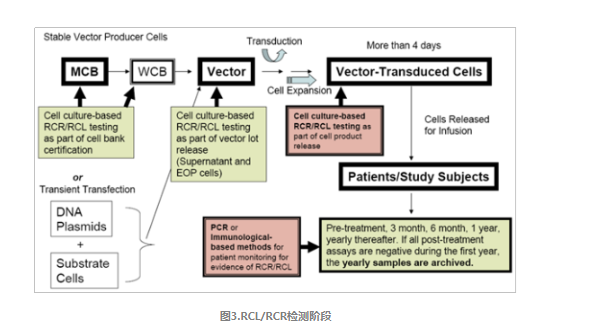
美国FDA要求对于采用γ-逆转录病毒载体和慢病毒载体的产品,需要在整个生产过程及不同阶段进行RCL/RCR检测,针对载体生产用主细胞库、工作细胞库、生产终末细胞、载体上清液及在体外培养超过4天的载体转到细胞进行检测,具体如如图3。针对慢病毒载体使用时RCL的检测,除了金标准——指示细胞培养法,还需要选择2种不同原理的快检方法对其进行补充检测。
参考文献:
1、FDA. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs) Guidance for Industry[S]2020.
2、FDA. Testing of Retroviral Vector-Based Human Gene Therapy Products for Replication Competent Retrovirus During Product Manufacture and Patient Follow-up Guidance for Industry [S]. 2020.
3、Briefing Document — Testing for Replication Competent Retrovirus (RCR)/Lentivirus (RCL) in Retroviral and Lentiviral Vector Based Gene Therapy Products — Revisiting Current FDA Recommendations.
4、Guideline on quality, non-clinical and clinical aspects of medicinal products containing genetically modified cells. [S]. 2020
5、国家食品药品监督管理总局. 免疫细胞治疗产品药学研究与评价技术指导原则(试行). 2022.
6、《体内基因治疗产品药学研究与评价技术指导原则(试行)》,国家药监局药品审评中心,2022.
7、《免疫细胞治疗产品药学研究与评价技术指导原则(试行)》,国家药监局药品审评中心,2022.
8、《体外基因修饰系统药学研究与评价技术指导原则(试行)》,国家药监局药品审评中心,2022.
9、《体外基因修饰系统药学研究与评价技术指导原则(试行)》,国家药监局药品审评中心,2022.
10、卢加琪, 李倩与何伍, 《体外基因修饰系统药学研究与评价技术指导原则(试行)》解读. 中国新药杂志, 2023. 32(02): 第118-122页.
<上下滑动查看更多>