GDC-1971——SHP2抑制剂引领癌症治疗新纪元

简介
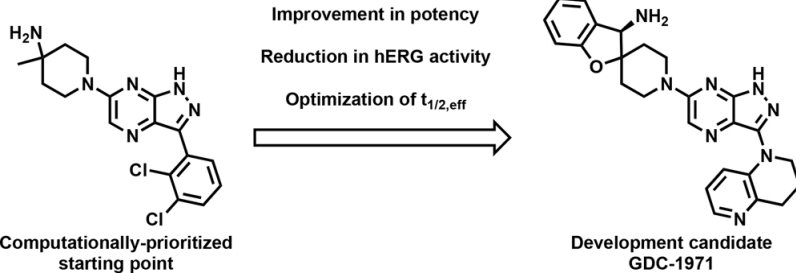
近年来,蛋白酪氨酸磷酸酶SHP2在介导RAS驱动的MAPK信号转导过程中,已成为肿瘤学领域备受瞩目的研究靶点。其治疗方法不仅限于单药使用,还能与KRAS抑制剂共同协作,展现出良好的治疗潜力。研究者初步发现,某些类似药物能够与异构位点结合,进而使酶保持在封闭、非活性的状态。特别值得一提的是,GDC-1971(原名RLY-1971)作为一种SHP2抑制剂,目前正在与KRAS G12C抑制剂divarasib(GDC-6036)联合应用于临床试验中,共同对抗由KRAS G12C突变驱动的实体瘤,为肿瘤治疗领域带来了新的希望。
GDC-1971的合成
在药物化学的优化历程中,GDC-1971等化合物主要遵循两条合成路径。这两条路径均可采用相似的化学技术,针对核心结构、碱性胺和四氢萘啶区域进行调整,进而将多样化的结构单元有效结合,从而实现化合物的优化与改良。
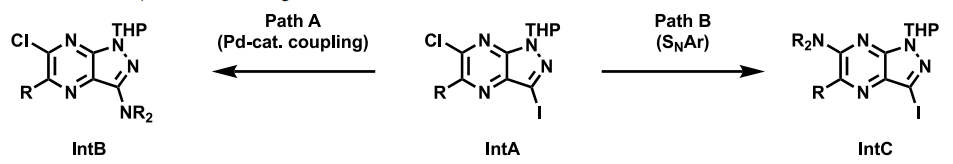
类似物的一般合成
从2,6-二氯吡嗪起始,可以通过对其进行石化处理,并使用甲酸乙酯进行淬灭反应,从而合成得到3,5-二氯吡嗪-2-甲醛。鉴于这种醛的化学性质较为不稳定,为了更有效地分离它,倾向于将其转化为相应的亚硫酸氢钠加合物形式。进一步地,当将此加合物与肼进行处理时,会生成一种加合物混合物。然而,在这过程中,相应的腙会发生分子内的环化反应,进而转化为6-氯-1H-吡唑并[3,4-b]吡嗪。随后,通过碘化过程,获得了化合物44。为了得到所需的氯碘中间体45,接着采用二氢吡喃进行保护处理。
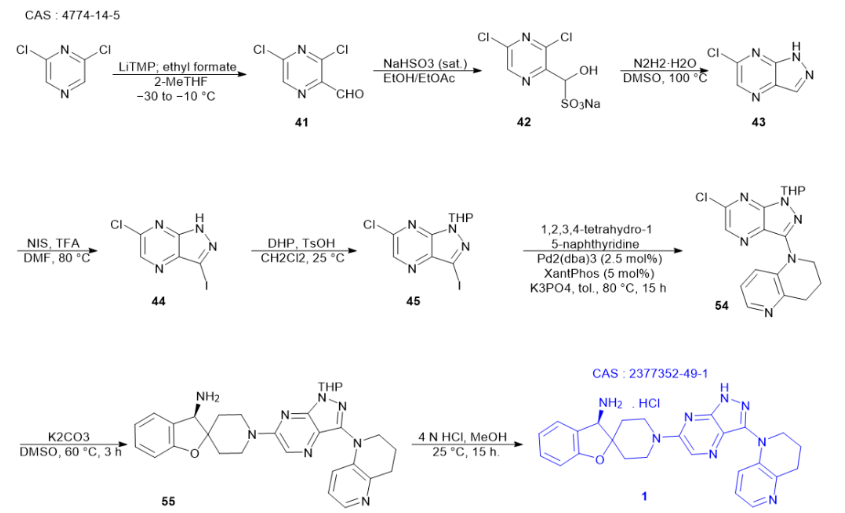
在碱性环境下,关键中间体1-(6-氯-1-(四氢-2H-吡喃-2-基)-1H-吡唑并[3,4-b]吡嗪-3-基)-1,2,3,4-四氢-1经过一系列反应,首先与四氢萘啶和Xantphos通过钯催化偶联作用,生成了5-萘啶。随后,这个杂芳基氯在二甲基甲酰胺中与(R)-3H-螺[苯并呋喃-2,4′-哌啶]-3-胺的双盐酸盐和二异丙基乙胺发生亲核芳香取代反应,最终得到GDC-1971的四氢吡喃保护类似物。为了进一步处理,这个盐可以用氢氧化钠水溶液进行游离,随后用2-甲基四氢呋喃进行萃取。这一系列步骤精细而关键,为GDC-1971的制备提供了重要的中间体。

芳基螺旋环胺构建块的代表性合成
以下方案详细描述了用于制备类似物的芳基螺环胺结构单元的合成过程。在这一过程中,首先利用埃尔曼助剂进行不对称还原反应,随后进行全局脱保护步骤,从而得到二胺盐。这一产物可以直接应用于如IntA等化合物的亲核芳香取代反应中,为后续的合成步骤提供了关键的原料。

GDC-1971中螺苯并呋喃哌啶环的合成
(R)-3H-螺[苯并呋喃-2,4′-哌啶]-3-胺,作为GDC-1971的关键碱性胺成分,其合成起始于2-氟苯甲醛。随后,通过一系列转化步骤,成功将其转化为相应的1,3-二噻烷,为后续的合成反应奠定了坚实基础。
GDC-1971的药理研究
GDC-1971,即(R)-1′-(3-(3,4-二氢-1,5-萘啶-1(2H)-基)-1H-吡唑并[3,4-b]吡嗪-6-基)-3H-螺[苯并呋喃-2,4′-哌啶]-3-胺,在生化及细胞环境中均展现出了对SHP2的显著抑制效果。

GDC-1971的选择参数:结构、理化和效价
为了更精准地预测GDC-1971在人体内的药代动力学(PK)特性及有效剂量,在临床前物种中进行了PK评估,并收集了临床前物种及人体系统中的体外ADME数据,以提供全面的药物代谢和动力学信息。

GDC-1971的多物种药动学特征
GDC-1971的体外与体内药效、药代动力学(PK)以及非靶向活性数据均显示,该药物能够在适宜的剂量范围内,以可接受的安全系数有效抑制人体内的SHP2。目前,该化合物已顺利进入进一步的安全性和IND使能研究阶段,并有望最终进入临床试验。关于其后续的研究进展和临床试验结果,将在适当时机予以披露。

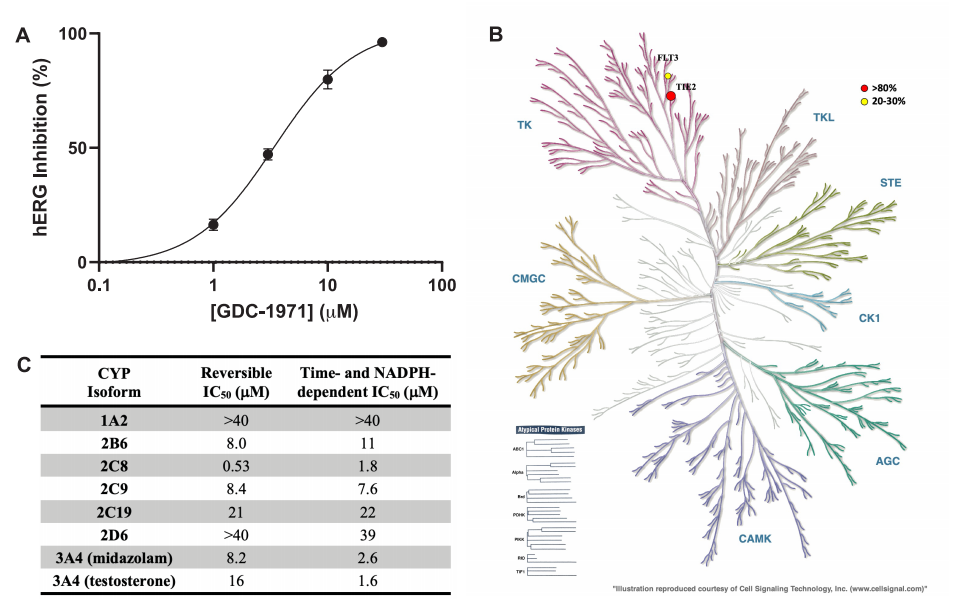
GDC-1971脱靶谱:(A)功能性hERG活性的剂量-反应曲线。(B)树突图显示一组激酶受到抑制。(C) CYP抑制谱。
结语
综上所述,GDC-1971在体外和体内临床前阶段所展现的特性,使其具备成为测试SHP2抑制作用的临床试验候选药物的巨大潜力。依据本文详述的合成路线,GDC-1971能够利用批量制备的构建模块实现大规模合成,为后续的临床研究提供了坚实的基础。
参考文献
1. Alexander M. Taylor. et al. Identification of GDC-1971 (RLY-1971), a SHP2 Inhibitor Designed for the Treatment of Solid Tumors. J. Med. Chem. 2023, 66, 19, 13384-13399. https://doi.org/10.1021/acs.jmedchem.3c00483