细菌基因组de novo测序
产品名称: 细菌基因组de novo测序
英文名称: Bacterial genome de novo sequencing
产品编号:
产品价格: 询价
产品产地: null
品牌商标: null
更新时间: null
使用范围: null
- 联系人 :
- 地址 : 上海市浦东新区国际医学园区康新公路3399弄3号楼
- 邮编 : 201203
- 所在区域 : 上海
- 电话 : 159****9102 点击查看
- 传真 : 点击查看
- 邮箱 : marketing@majorbio.com
产品介绍
随着高通量测序技术和生物信息学的快速发展,细菌全基因组denovo测序已然成为一种探究细菌生物学问题的高性价比方法。通过全基因组测序,可以获知待测菌株的基因及相关调控信息,为研究该菌株特有的生物学特征(致病机制,共生机制,独有的代谢机制)提供分子基础;通过比较基因组分析,可以研究种内及种间的相互进化关系,为探究疾病的传播机制提供理论指导。
目前,细菌基因组测序已经广泛应用于细菌流行病学、疫苗开发、微生物进化等多个领域。此外,细菌全基因组de novo测序也能为该物种后续基因组数据的挖掘以及重要基因的功能分析构建一个全面的研究平台。
美吉优势
拥有标准化操作实验室和高通量测序技术平台,实验周期短,保证高质量的数据;
拥有多种测序平台和强大的计算机资源,提供最佳的测序解决方案和快速的分析平台;
拥有专业的分析团队,全面的分析内容,提供全面和高质量的基因组解析结果;
产品类型
细菌基因组框架图
构建Illumina PE(~400bp)文库,通过Illumina(Miseq/Hiseq)平台测序并进行de novo基因组组装,初步获得基因组序列和功能注释信息。
细菌基因组完成图
构建PacBio RS II(~10K)文库,基于多种测序平台(ABI3730xl,Illumina Miseq/Hiseq,Pacbio RS II)获得基因组完成图并进行深度生物信息挖掘。
实验流程
框架图分析流程: 完成图实验流程: 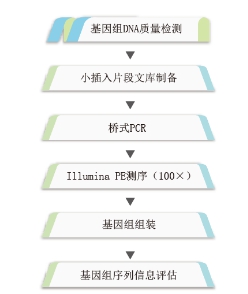
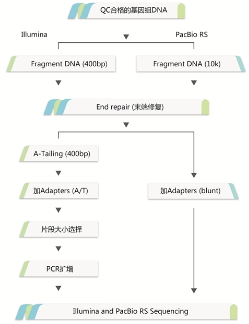
生信分析
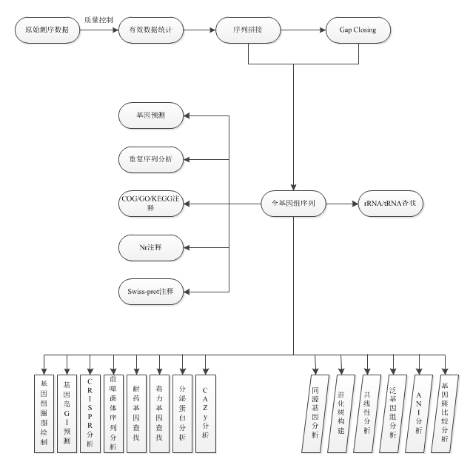
生信分析内容:
|
高级分析I(序列分析) |
高级分析II(比较基因组) | |
|
原始数据统计与处理 |
基因圈图绘制 |
同源基因分析 |
|
基因组拼接 |
基因岛(GIs)预测 |
基因组进化树构建 |
|
基因预测 |
CRISPR分析 |
共线性分析 |
|
重复序列分析 |
前噬菌体序列分析 |
泛基因组分析 |
|
非编码RNA预测 |
耐药和毒力基因注释 |
ANI分析 |
|
Nr/Swiss-prot注释 |
分泌蛋白分析 |
基因簇比较作图分析 |
|
COG/GO/KEGG注释 |
碳水化合物相关酶数据库(CAZy)注释 |
技术指标
框架图承诺指标:
基因组覆盖度大于95%,基因区覆盖度98%以上,单碱基错误率低于十万分之一
完成图承诺指标:
0 GAP,1 contig。
送样要求:
1)样品类型: DNA/菌体
2)样品需求量(单次):菌体样本:≥5g; 基因组DNA:≥2μg(框架图),基因组DNA:≥10μg(完成图)
3)样品浓度:≥20ng/µL
4)样品纯度:OD260/280=1.8-2.0并确保DNA无降解,无污染。
5)样品保存期间切忌反复冻融,送样时请使用冰袋或干冰运输。
案例解读
案例一:基因组分析揭示了Bacillus cereus Group中各菌株的分类地位[1]
本文利用224株芽孢杆菌基因组序列,通过GBDP/dDDH、16S rRNA以及特殊基因MLSA等方法进行芽孢杆菌分类地位研究。解决几个问题:1)比较这些方法的分类结果;2)重新构建蜡样芽孢杆菌的系统进化树;3)探索芽孢杆菌的遗传多样性;4)获得更满意的分类结果。
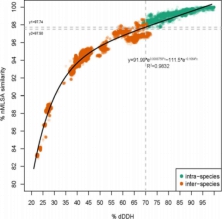
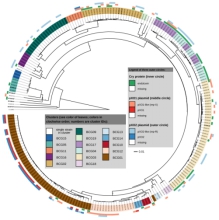
图1 dDDH值与nMLSA相似性的相关性分析 图2 pycA基因构建系统进化树
案例二:213株乳杆菌的比较基因组学分析[2]
本文对213株乳杆菌属模式菌株,采用二代Illumina HiSeq 2000高通量测序平台绘制其基因组精细图谱,结合比较基因组和功能基因组分析方法,解析与菌株分化相伴随的功能基因进化历程,拓展其生物应用潜力。
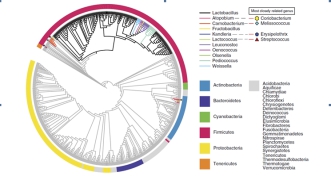

图1基于16个持家基因的最大似然法系统进化树 图2基于73个核心基因的系统进化树
参考文献
[1] Liu Y, Lai Q, Göker M, et al. Genomic insights into the taxonomic status of the Bacillus cereus group. Scientific Reports, 2015, 5: 14082-14082.
[2] Sun Z, Harris H M B, McCann A, et al. Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nature Communications, 2015, 6: 8322.
常见问题
(1)细菌基因组组装效果的指标有那些?在一个什么样的范围算是比较正常的?
我们进行多个k-mer参数的拼接后,会综合考虑scaffold N50长度值、N%、scaffold数量、总碱基数等指标,一般要求N50较长,N%较低,scaffold数量相对较少,这样拼接比较集中,不至于太零散;依据这些技术指标进行综合评定,来选取最终的组装结果;因为不同菌的基因组有差异,分析要达到的精细程度也不同,所以会根据客户要求和基因组情况选取最佳结果,各个指标的范围也不同。
(2)对于GC含量过高或过低以及重复序列较多的基因组,完成图能否0Gap?
GC含量大于65%或者小于35%以及重复序列较多时,会对测序和组装有一定影响,但目前Pacbio长读长的测序技术能很好的克服这些问题,美吉生物有几百例的细菌完成图项目经验,利用三代测序技术全部都可以达到0gap的结果。