简化基因组测序
产品名称: 简化基因组测序
英文名称: Reduced-Representation Genome Sequencing
产品编号:
产品价格: 0
产品产地: null
品牌商标: null
更新时间: 2023-09-21T16:14:01
使用范围: null
- 联系人 :
- 地址 : 上海市徐汇区银都路218号聚科生物园区2号楼
- 邮编 : 200231
- 所在区域 : 上海
- 电话 : 186****3700 点击查看
- 传真 : 点击查看
- 邮箱 : market@personalbio.cn;lif@personalbio.cn
简化基因组测序(Reduced-Representation Genome Sequencing, RRGS)顾名思义是对部分基因组进行序列测定。它是指利用生物信息学方法,设计标记开发方案,筛选特异性长度片段,应用高通量测序技术获得海量标签序列来充分代表目标物种全基因组信息的测序方法。
RRGS技术在模式和非模式生物遗传图谱构建、系统进化、全基因组关联分析、基因或QTL定位和动植物分子育种等方面具有广泛的应用前景。
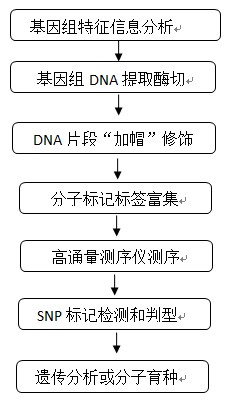
一、技术路线
推荐平台:
二、生物信息分析
1)未知参考基因组的信息分析流程
1. 原始数据整理、过滤及质量评估
2. 标记位点分析:
l 标记位点鉴定
l SNPs位点鉴定
3. 应用分析:
l SNP单体型图谱分析
l 全基因组关联分析(GWAS)
l 连锁分析和QTL定位
l 种群进化分析
4. 根据客户需求进行个性化分析
2)已知参考基因组的信息分析流程
1. 原始数据整理、过滤及质量评估
2. 标记位点分析:
l 标记位点在基因组上定位
l 标记位点鉴定
l SNPs位点鉴定
3. 应用分析:
l SNP单体型图谱分析
l 全基因组关联分析(GWAS)
l 连锁分析和QTL定位
l 种群进化分析
4. 根据客户需求进行个性化分析
三、样品要求
1. 动物组织样品:要求新鲜动物组织,避免脂肪组织,每个样品重量>10 g。
2. 血液样品:每份新鲜抗凝血液样本的体积>5 mL。
3. 植物样品:要求新鲜较嫩的叶片组织,每个样品重量>10 g,干冰或者液氮保存寄送。
4. DNA样品:请提供浓度>100 ng/μl,总量>20 μg的DNA,OD260/280介于1.8-2.0之间,并确保DNA无降解。
5. 样品保存期间切忌反复冻融。
6. 送样管务必标清样品编号,管口使用Parafilm膜密封。
四、经典案例
案例一 群体SNP遗传图谱构建和进化分析
背景:瓶草蚊(pitcher plant mosquito)是第一个被发现在遗传学上适应气候快速变化的物种。它不是模式生物,也没有基因组的参考序列。
目的:采用RAD-Seq(restriction-site-associated DNA sequencing)方法探索不同地理分布瓶草 蚊群体进化关系。

结果:对北美21个不同地理分布的瓶草蚊群体进行分析,使用8个碱基识别位点的内切酶SbfI消化gDNA,在13627个位点上获得了3741 SNPs。使用最大似然法成功地将21个群体区分为4个大分支,而且21个群体间的进化关系得到的阐明。
[ Emerson KJ, Merz CR, Catchen JM, et al. Resolving postglacial phylogeography using high-throughput sequencing. Proc Natl Acad Sci, 2010, 107: 16196-16200 ]
案例2:性状-标记全基因组关联分析(GWAS)
背景:水稻是重要的粮食作物,了解农业性状决定基因所在区域或位点对遗传机制研究和分子育种有重要意义。 目的:构建水稻高密度单倍型图谱,并对部分农业性状进行了全基因组关联分析(GWAS)。 结果:采用GBS方法对517个水稻地方品种测序,获得360万个SNP位点。针对14种农业性状,如表型特征、产品构成、籽实质量、颜色和生理特性等进行了GWAS。结果发现了36%表型变异同SNP位点关联,其中6个位点靠近先前已知关联基因,标记分辨率在不小于27 kb。证明了这种方法可用于高密度单倍型图谱构建和GWAS研究。 [ Huang XH, Wei XH, Sang T, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nature Genetics. 2010, 42: 961–967 ] 五、常见问题和解答 1. Q:高密度SNPs标记开发时要考虑哪些因素? A:高密度SNPs标记开发的基本条件和设计理念是所获得的片段尽量在基因组上分布均匀,通过测定少量代表全基因组信息的序列获得的成千上万SNPs标记。首先要利用生物信息学方法对目标物种参考基因组(或已知BAC序列)进行系统分析,根据基因组GC含量、重复序列情况和基因特点等信息,选择相应的限制性酶以及测序文库类型,以保证分子标记的密度、均匀性、效率满足遗传分析和分子育种的需要。 2. Q:简化基因组测序的常用测序文库类型有哪些? A:简化基因组测序常用测序文库类型可以归纳为3个大类,①简化测序,包括简化文库(reduced-representation libraries,RRLs)和简化多态序列复杂性(complexity reduction of polymorphic sequences,CroPS);②限制性酶切位点关联DNA测序(restriction-site-associated DNA sequencing,RAD-seq);③低覆盖基因分型文库,包括多元鸟枪法基因分型(multiplexed shotgun genotyping,MSG)和基于测序的基因分型(genotyping by sequencing,GBS)。