α-突触核蛋白与帕金森氏症的病因与疾病发展有着密切的联系。帕金森氏症 (PD) 是一种神经衰退疾病,该疾病患者的大脑中可见α-突触核蛋白聚集形成路易体。α-突触核蛋白是由SNCA基因表达的一种分子量为 14-kDa 的蛋白。目前已知的α-突触核蛋白的自然状态可有很多种:可能是没有折叠的单体1、折叠的四聚体、或者是处于和其他寡聚体动态共存的形式2。在帕金森疾病中,这些分子量小的α-突触核蛋白会聚集形成原纤维、纤维以及路易体,从而导致神经元的病变和死亡。也有其他研究表明寡聚体和原纤维具有神经毒性3-6而路易小体可能有神经保护功能7。
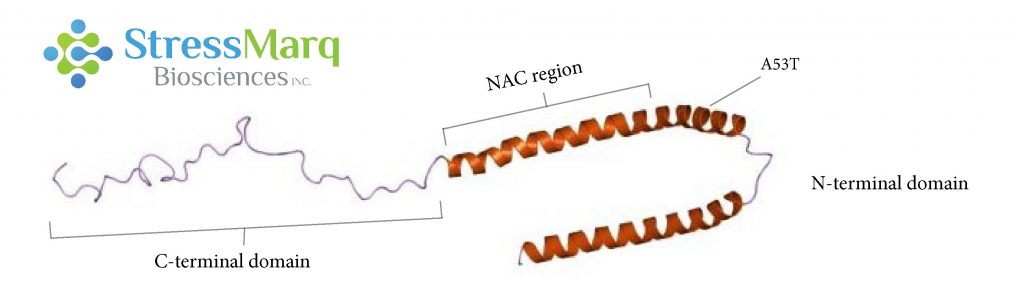
α-突触核蛋白由三个主要区域:羧基末端区域、NAC区域、以及氨基末端区域。A53T 突变位于氨基末端区域。
A53T 是错义点突变, 也就是导致了氨基酸的改变:第53 个氨基酸由丙氨酸突变为苏氨酸。该突变是由于SNCA基因 209位置的鸟嘌呤变成了腺嘌呤 (G209A)8。A53T 突变与一种常染色体显性遗传的早发型 PD 相关,这种 PD 最先发现于意大利和希腊裔家族8,也见于一个韩国裔家族9。 A53T 变异导致该疾病的发病年龄比较早8。虽然大部分的 PD 病例是散发型的,并非遗传病,而且也不涉及 A53T 突变, 但是研究 A53T 突变可以帮助科研学者们更好的了解α-突触核蛋白的聚集和发展机制,从而研发出更好的疾病模型和治疗方案。
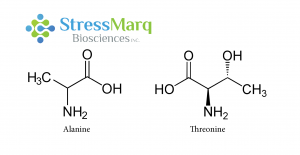
丙氨酸和苏氨酸有相似的结构,但是α-突触核蛋白中的丙氨酸替换成苏氨酸对α突触核蛋白的纤维原聚集有很大的作用。
A53T 变异只涉及到单一氨基酸改变,而且丙氨酸和苏氨酸结构上非常相近,那么为什么A53T突变型有这么强的聚集效果呢?这是因为A53T 和其他致病突变都发生在α-突触核蛋白的氨基末端,理论模型显示 A53T 突变会使α-突触核蛋白的 NAC 区域和氨基或羧基区域的长程相互作用消失,导致 beta折叠的加速形成10。NMR 测量数据显示 A53T 突变可以延长并稳定在寡聚化和纤维化中起着重要作用的 beta折叠结构11。因此A53T 突变型能够更快聚集alpha突触核蛋白,有更明显的致病效果。下面的硫黄素T检测曲线可以明显看出A53T突变型的聚集效果明显强于非突变型α-突触核蛋白。
A53T 变异只涉及到单一氨基酸改变,而且丙氨酸和苏氨酸结构上非常相近,那么为什么A53T突变型有这么强的聚集效果呢?这是因为A53T 和其他致病突变都发生在α-突触核蛋白的氨基末端,理论模型显示 A53T 突变会使α-突触核蛋白的 NAC 区域和氨基或羧基区域的长程相互作用消失,导致 beta折叠的加速形成10。NMR 测量数据显示 A53T 突变可以延长并稳定在寡聚化和纤维化中起着重要作用的 beta折叠结构11。因此A53T 突变型能够更快聚集alpha突触核蛋白,有更明显的致病效果。下面的硫黄素T检测曲线可以明显看出A53T突变型的聚集效果明显强于非突变型α-突触核蛋白。
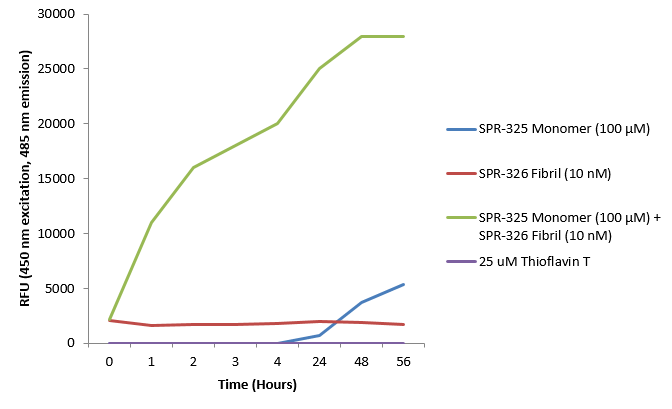 |
硫黄素 T 是一种荧光染料, 可以绑定富含beta折叠的结构, 例如 α-Syn 聚合体. 绑定之后, 硫黄素T光谱会发生红移, 荧光强度会增强. A53T α-Syn蛋白单体 (SPR-325)的硫黄素 T 发射光曲线显示了有限的荧光强度增强 (相关于α-Syn 蛋白聚合体). 而 10 nM 活性 α-Syn 聚合体 (SPR-326) 与100 μm 活性 α-Syn 单体 (SPR-325) 混合时荧光强度明显增强, 这是由于聚合体催化了活性单体形成更多聚集体PFFs. Thioflavin T λex = 450 nm, λem = 485 nm. |
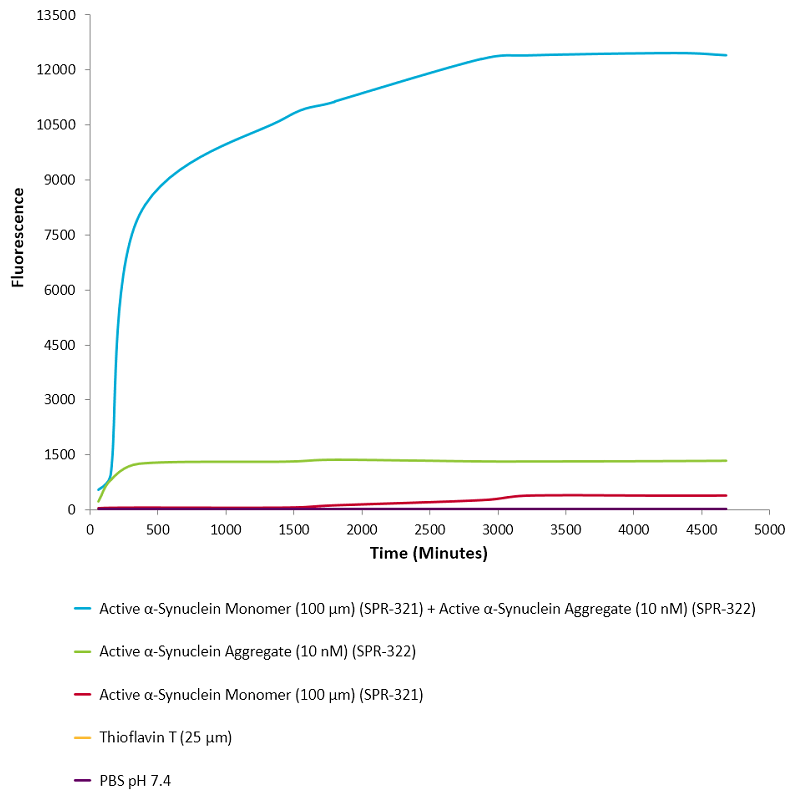 |
活性突触核蛋白 (α-Syn) 聚合体 (SPR-322) 催化活性单体(SPR-321)形成新的聚合体. 硫黄素 T 是一种荧光染料, 可以绑定富含beta折叠的结构, 例如 α-Syn 聚合体. 绑定之后, 硫黄素T光谱会发生红移, 荧光强度会增强. 左侧硫黄素 T 发射光曲线展示了四种试验对象随时间增强的荧光强度(相关于α-Syn 蛋白聚合体),其中 10 nM 活性 α-Syn 聚合体(SPR-322) 与100 μm 活性 α-Syn 单体 (SPR-321) 混合物荧光增强最明显, 另外几组对比分别是活性 α-Syn 聚合物 (SPR-322) 、活性 α-Syn 单体 (SPR-321) 以及 Thioflavin T λex = 450 nm, λem = 485 nm. |
A35T突变型 alpha 突触核蛋白新品上市半价促销,活动时间2019年3月1日至4月30日,折扣码 A35T50.
| 活性重组人 A53T 突变型 Alpha Synuclein 蛋白单体 (1 型) | SPR-325B | 100 µg |
| SPR-325C | 2 x 100 µg | |
| SPR-325E | 5 x 100 µg | |
| 活性重组人 A53T 突变型 Alpha Synuclein 蛋白 PFFs (1 型) | SPR-326B | 100 µg |
| SPR-326C | 2 x 100 µg | |
| SPR-326E | 5 x 100 µg |
参考文献:
- Fauvet B, et al. alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287:15345–64.
- Dehay B, Bourdenx M, Gorry P, et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 2015;14(8):855-866.
- Karpinar DP, et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 2009;28:3256–68.
- Winner B, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–9.
- Cremades N, et al. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. 2012;149:1048–59.
- Danzer KM, et al. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci. 2007;27:9220–32.
- Tanaka M, et al. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem. 2004;279:4625–31.
- Polymeropoulos, M. H. Mutation in the -Synuclein Gene Identified in Families with Parkinson’s Disease. Science, 1998;276(5321), 2045–2047. doi:10.1126/science.276.5321.2045
- Ki C.S. Stavrou E.F. Davanos N. Lee W.Y. Chung E.J. Kim J.Y. Athanassiadou A. The Ala53Thr mutation in the alpha-synuclein gene in a Korean family with Parkinson disease. Clin Genet. 2007 May;71(5):471-3.
- Coskuner, O., Wise-Scira, O. Structures and Free Energy Landscapes of the A53T Mutant-Type α‑Synuclein Protein and Impact of A53T Mutation on the Structures of the Wild-Type α‑Synuclein Protein with Dynamics. ACS Chem. Neurosci. 2013, 4, 1101−
- Russel, R., Eliezer, D. Residual structure and dynamics in Parkinson’s disease-associated mutants of alpha-synuclein. J Biol Chem. 2001 Dec 7;276(49):45996-6003.